
 电动病床CE认证,矫形眼镜CE认证、矫形架CE认证、诊断用听诊器CE认证
电动病床CE认证,矫形眼镜CE认证、矫形架CE认证、诊断用听诊器CE认证
 HAD-YVT-2 机械故障听诊器 工业听诊器 设备故障听诊器 机械听诊器
HAD-YVT-2 机械故障听诊器 工业听诊器 设备故障听诊器 机械听诊器
 HAD-YVT-2 机械故障听诊器 工业听诊器 设备故障听诊器 机械听诊器
HAD-YVT-2 机械故障听诊器 工业听诊器 设备故障听诊器 机械听诊器
 HAD-YVT-2 机械故障听诊器 工业听诊器 设备故障听诊器 机械听诊器
HAD-YVT-2 机械故障听诊器 工业听诊器 设备故障听诊器 机械听诊器
 DP-YVT-2 机械故障听诊器 工业听诊器 设备故障听诊器 机械听诊器
DP-YVT-2 机械故障听诊器 工业听诊器 设备故障听诊器 机械听诊器
 XRS-YVT-2 机械故障听诊器 工业听诊器 设备故障听诊器 机械听诊器
XRS-YVT-2 机械故障听诊器 工业听诊器 设备故障听诊器 机械听诊器
 北京机械状态听诊器,机械故障听诊器,北京机械状态听诊器,机械故障听诊器
北京机械状态听诊器,机械故障听诊器,北京机械状态听诊器,机械故障听诊器
 供应机械状态听诊器,机械故障听诊器,北京卓川机械状态听诊器,机械故障听诊器
供应机械状态听诊器,机械故障听诊器,北京卓川机械状态听诊器,机械故障听诊器
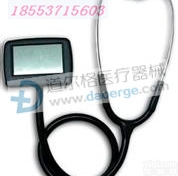 DEG8547 厂家直销外销听诊器 平面听诊器 单面 单头听诊器医用 医用听诊器
DEG8547 厂家直销外销听诊器 平面听诊器 单面 单头听诊器医用 医用听诊器
 HAD-YVT-2 机械故障听诊器 工业听诊器 设备故障听诊器 机械听诊器.
HAD-YVT-2 机械故障听诊器 工业听诊器 设备故障听诊器 机械听诊器.
 DP-YVT-2 机械故障听诊器 工业听诊器 设备故障听诊器 机械听诊器
DP-YVT-2 机械故障听诊器 工业听诊器 设备故障听诊器 机械听诊器
 机器故障电子听诊器 便携式故障电子听诊器 故障电子听诊器
机器故障电子听诊器 便携式故障电子听诊器 故障电子听诊器
本产品信息由(上海真科产品检测技术有限公司)为您提供,内容包括(电动病床CE认证,矫形眼镜CE认证、矫形架CE认证、诊断用听诊器CE认证)的品牌、型号、技术参数、详细介绍等;如果您想了解更多关于(电动病床CE认证,矫形眼镜CE认证、矫形架CE认证、诊断用听诊器CE认证)的信息,请直接联系供应商,给供应商留言。若当前页面内容侵犯到您的权益,请及时告知我们,我们将马上修改或删除。

沪公网安备 31011502008050号


