
提供商: illumina



1. 原始测序数据
高通量测序(如illumina HiSeqTM X 10等测序平台)测序得到的原始图像数据文件经碱基识别(Base Calling)分析转化为测序序列(Sequenced Reads),我们称之为Raw Data或Raw Reads,结果以FASTQ(简称为fq)文件格式存储,其中包含测序序列(reads)的序列信息以及其对应的测序质量信息。
FASTQ格式文件中每个read由四行描述,如下:
@HWI-ST1276:71:C1162ACXX:1:1101:1208:2458 1:N:0:CGATGT
NAAGAACACGTTCGGTCACCTCAGCACACTTGTGAATGTCATGGGATCCAT
+
#55???BBBBB?BA@DEEFFCFFHHFFCFFHHHHHHHFAE0ECFFD/AEHH
其中DY行以“@”开头,随后为Illumina 测序标识别符(Sequence Identifiers)和描述文字(选择性部分);第二行是碱基序列;第三行以“+”开头,随后为 illumina 测序标识符(选择性部分);第四行是对应序列的测序质量(Cock et al.)。
Illumina测序标识符详细信息如下:

第四行中每个字符对应的ASCII值减去33,即为对应第二行碱基的测序质量值。如果测序错误率用e表示,illumina HiSeqTM X 10的碱基质量值用Qphred表示,则有下列关系:
公式一: Qphred = -10log10(e)
illumina Casava 1.8版本测序错误率与测序质量值简明对应关系如下:

2 测序数据质量评估
2.1 测序错误率分布检查
每个碱基测序错误率是通过测序Phred数值(Phred score,Qphred)通过公式1转化得到,而Phred 数值是在碱基识别(Base Calling)过程中通过一种预测碱基判别发生错误概率模型计算得到的,对应关系如下表所显示。
illumina Casava 1.8版本碱基识别与Phred分值之间的简明对应关系如下表:

测序错误率与碱基质量有关,受测序仪本身、测序试剂、样品等多个因素共同影响。对于RNA-seq技术,测序错误率分布具有两个特点:
(1) 测序错误率会随着测序序列(Sequenced Reads)长度的增加而升高,这是由于测序过程中化学试剂的消耗而导致的,并且为Illumina高通量测序平台都具有的特征(Erlich and Mitra, 2008; Jiang et al., 2011)。
(2) 前几个碱基的位置也会发生较高的测序错误率,是因为在RNA-seq建库过程中反转录需要随机引物,推测前几个碱基测序错误率较高的原因是随机引物碱基和RNA模版的不完全结合(Jiang et al., 2011)。
测序错误率分布检查用于检测在测序长度范围内,有无测序错误率异常的碱基位置。一般情况下,每个碱基位置的测序错误率都应该低于0.5%。项目结果见下图:

图2.1 测序错误率分布图
横坐标为reads的碱基位置,纵坐标为单碱基错误率
2.2 测序数据质量情况汇总
样品测序产出原始数据质量评估情况详见表 2.1。
表 2.1 数据产出质量情况一览表

注:
(1) Sample:样品id。
(2) Raw Reads:统计原始序列数据,以四行为一个单位,统计每个测序文件的测序序列的个数。
(3) Clean Reads:经过过滤后的数据。
(4) Clean Bases:测序序列的个数乘以测序序列的长度,并转化为以G为单位。
(5) Error rate:指测序错误率,通过公式一计算得到。
(6) Q20:Phred数值大于20的碱基占总体碱基的百分比。
(7) Q30:Phred数值大于30的碱基占总体碱基的百分比。
(8) GC content:计算碱基G和C的数量总和占总体碱基数量的百分比。
3 circRNA 的鉴定
使用 find_circ (Sebastian Memczak et al., 2013) 鉴定 circRNA。find_circ 的基本原理是从没有比对到参考序列的 reads 的两端各提取 20-nt 的 anchor 序列,将每一对 anchor 序列再次比对参考序列,如果 anchor 序列的 5' 端比对到参考序列(起始与终止位点分别记为 A3,A4),同时该 anchor 序列的 3' 端比对到此位点的上游(起始与终止位点分别记为 A1,A2),并且在参考序列的 A2 到 A3 之间存在剪接位点(GT-AG),则将此 read 作为候选 circRNA。ZH将 read count 大于等于 2 的候选 circRNA 作为鉴定的 circRNA。
circRNA 鉴定的部分结果见下表:

注:
(1) circRNA_id: circRNA ID(此 ID 与每个样品中的 circRNA ID 不同)
(2) chr: 染色体编号
(3) start: circRNA 全长的起始位点
(4) end: circRNA 全长的终止位点
(5) strand: 正负链
(6) full_length: circRNA 全长
3.1 circRNA 的长度分布
所有样本的 circRNA 的长度分布见下图:

图 3.1 circRNA 的长度分布
3.2 circRNA 的来源统计
circRNA 可以来源于 exon 或 intron 的剪接,下图统计了所有样本的 circRNA 的来源

图 3.2 circRNA 的来源
3.3 已知 circRNA 的注释
如果样品物种存于 circBase 中,将所有鉴定的 circRNA 与 circBase 中该物种已知 circRNA 比较,统计测序结果中已知 circRNA 的数量及比例。


图 3.3 鉴定的 circRNA 与 circBase 已知 circRNA 的比较
柱状图:对于每个样品,分别统计鉴定的 circRNA 中存在于 circBase 中的 circRNA 的数量(起始位点及终止位点的 overlap 均不超过 2-nt);
饼图:统计所有鉴定的 circRNA 中,存在于 circBase 中的 circRNA 的比例。
4 circRNA 的表达及差异表达
4.1 circRNA 的表达水平分析
对各样本中已知和新 circRNA 进行表达量的统计,并用 TPM(Zhou et al., 2010)进行表达量归一化处理。
公式:归一化表达量 = (readCount*1,000,000)/libsize (libsize:样品 circRNA readcount 之和)
表4.1 circRNA表达水平统计表(部分)

注:结题报告中只显示部分circRNA表达水平统计结果,完整信息见结果文件夹。
(1) 第1列——“circRNA” ID。
(2) 第2到n+1列——“样本名”,分别代表样本1到n的readcount。
(3) 第 n+2 到 2n+1 列——“样本名(TPM)”,分别代表样本 1 到 n 的 readcount 经过 TPM 标准化后的值。
4.2 circRNA表达量TPM密度分布图
TPM密度分布能整体检查样品的基因表达模式,项目结果见图4.1。

图 4.1 TPM密度分布图
横坐标为circRNA的log10(TPM+1)值,纵坐标为对应log10(TPM+1)的密度
4.3 circRNA差异表达分析结果
circRNA差异表达的输入数据为circRNA表达水平分析中得到的readcount数据。对于有生物学重复的样品,分析我们采用基于负二项分布的DESeq2(Michael et al., 2014)进行分析;对于无生物学重复的样品,先采用TMM对readcount数据进行标准化处理,之后用DEGseq(Wang et al., 2010)进行差异分析。
差异分析的部分结果示意见表4.2。

注:
(1) circRNA_ID: circRNA ID。
(2) Group1:校正后样品1的read count值。
(3) Group2:校正后样品1的read count值。
(4) log2.Fold_change.:log2(Sample1/Sample2)。
(5) p.value:p值
4.4 差异circRNA筛选
用火山图可以推断差异circRNA的整体分布情况,从差异倍数(Fold change)和校正后的显著水平(padj/qvalue)两个水平进行评估,对差异circRNA进行筛选。
当样品有生物学重复时,默认差异circRNA的筛选条件为:padj<0.05。
当样品无生物学重复时,差异circRNA数目会偏多,为了控制假阳性率,需qvalue结合foldchange来筛选,默认差异circRNA筛选条件为: qvalue<0.01 && | log2(foldchange)|>1。
项目结果见图4.3:

图4.3 差异circRNA火山图
横坐标代表circRNA在不同实验组中/不同样品中表达倍数变化,纵坐标代表circRNA表达量变化的统计学显著程度,图中的散点代表各个circRNA,蓝色圆点表示无显著性差异的circRNA,红色圆点表示显著上调的差异circRNA,绿色圆点表示显著下调的差异circRNA
4.5 差异circRNA聚类分析
差异circRNA聚类分析用于判断不同实验条件下差异circRNA表达量的聚类模式。 每个比较组合都会得到一个差异circRNA集,将所有比较组合的差异circRNA集的并集在每个实验组/样品中的的TPM值,用于层次聚类分析(结果见图4.4),K-means聚类分析和SOM聚类分析(见结果文件夹)。

图4.4 差异circRNA聚类图
上图为整体层次聚类图,以log10(TPM+1)值进行聚类,红色表示高表达circRNA,蓝色表示低表达circRNA
4.6 样本间公共及特有的 circRNA 维恩图
当比较组合数大于等于 2 个小于 5 个时,可以将各组比较得到的差异 circRNA 个数进行统计,画成维恩图,直观展现出各个比较组合共有的及特有的差异 circRNA 数目(如果只有两个样品时,则进行两个样本之间的venn 图绘制)(见图4.5)。

图4.5 差异circRNA维恩图
大圆圈代表各个比较组合,每个大圆圈中的数字之和代表该比较组合的差异circRNA总个数,圆圈交叠的部分表示组合之间共有的差异circRNA个数(只有两个样品时,每个大圆圈中的数字代表在这个样本中表达的circRNA,圆圈交叠的部分代表样本间共表达的circRNA)
5 差异 circRNA 来源基因富集分析
得到各组比较间的差异表达 circRNA 后,根据 circRNA 与其来源基因间的对应关系,对每组差异表达 circRNA 的来源基因的集合分别进行 Gene Ontology 和 KEGG 富集分析。
5.1 来源基因GO富集分析
Gene Ontology(简称 GO,http://www.geneontology.org/)是基因功能国际标准分类体系。GO 富集分析方法为 GO seq ,此方法基于Wallenius non-central hyper-geometric distribution,相对于普通的 Hyper-geometric distribution,此分布的特点是从某个类别中抽取个体的概率与从某个类别外抽取一个个体的概率是不同的,这种概率的不同是通过对基因长度的偏好性进行估计得到的,从而能更为准确地计算出 GO term 被来源基因富集的概率。结果如表 5.1所示。
表 5.1 样品中来源基因的 Gene Ontology 富集列表(部分)

注:
(1) GO accession: Gene Ontology 数据库中WY的标号信息。
(2) Description: Gene Ontology 功能的描述信息。
(3) Term type: 该 GO 的类别(cellular_component: 细胞组分;biological_process: 生物学过程;molecular_function: 分子功能)。
(4) Over represented p-value: 富集分析统计学显著水平。
(5) Corrected p-value: 校正后的 p-value,一般情况下,校正后的 p < 0.05 该功能为显著富集项。
(6) CAD item: 来源基因中与该 Term 相关的基因数。
(7) CAD list: GO 注释的来源基因数目
(8) Bg item: 背景(所有)基因中与该 Term 相关的基因数。
(9) Bg list: GO 注释的背景(所有)基因的数目
得到上述列表后,统计被显著富集的各个 GO term 中的基因数,以柱状图的形式展示,如图 5.1所示。

图5.1 来源基因 GO 富集柱状图
有向无环图(Directed Acyclic Graph,DAG)为来源基因 GO 富集分析结果的图形化展示方式,分支代表包含关系,从上至下所定义的功能范围越来越小,一般选取GO富集分析的结果前 10 位作为有向无环图的主节点,并通过包含关系,将相关联的 GO Term 一起展示,颜色的深浅代表富集程度。我们的项目中分别绘制生物过程(biological process)、分子功能(molecular function)和细胞组分(cellular component)的来源基因 DAG 图。

图 5.2 GO富集有向无环图
5.2 来源基因 KEGG 富集分析
在生物体内,不同基因相互协调行使其生物学功能,通过 Pathway 显著性富集能确定来源基因参与的最主要生化代谢途径和信号转导途径。KEGG(Kyoto Encyclopedia of Genes and Genomes)是有关 Pathway 的主要公共数据库(Kanehisa et al.,2008)。Pathway 显著性富集分析以KEGG Pathway 为单位,应用超几何检验,找出与整个基因组背景相比,在来源基因中显著性富集的 Pathway。该分析的计算公式:

其中,N 为所有基因中具有 Pathway 注释的基因数目;n 为 N 中来源基因的数目;M 为所有基因中注释为某特定 Pathway 的基因数目;m 为注释为某特定 Pathway 的来源基因数目。用 BH 方法对 p-value 进行校正,得到的校正后的 p-value 值越小代表越显著。这里将值小于 0.05 的 Pathway 定义为在来源基因中显著富集的 Pathway。
表 5.2 来源基因 KEGG 显著性富集列表

注:
(1) Term:KEGG 通路的描述信息。
(2) Id:KEGG 数据库中通路WY的编号信息。
(3) Sample number:在该通路下的来源基因数。
(4) Background number:在该通路下的背景基因数。
(5) P-value:富集分析统计学显著水平。
(6) Corrected p-value:校正后的统计学显著水平,一般情况下,校正后的 p-value < 0.05 该功能为显著富集项。
来源基因 KEGG 富集散点图是 KEGG 富集分析结果的图形化展示方式。在此图中,KEGG 富集程度通过 Rich factor、Qvalue 和富集到此通路上的基因个数来衡量。其中 Rich factor 指差异表达的基因中位于该 pathway 条目的基因数目与所有有注释基因中位于该 pathway 条目的基因总数的比值。Rich factor 越大,表示富集的程度越大。Qvalue 是做过多重假设检验校正之后的 p-value,Qvalue 的取值范围为 [0,1],越接近于 0 表示富集越显著。我们挑选了富集最显著的 20 条 pathway 条目在该图中进行展示,若富集的 pathway 条目不足 20 条,则全部展示。

图 5.3 来源基因 KEGG 富集散点图
纵轴表示 pathway 名称,横轴表示 Rich factor,点的大小表示此 pathway中来源基因个数多少,而点的颜色对应于不同的 Qvalue 范围
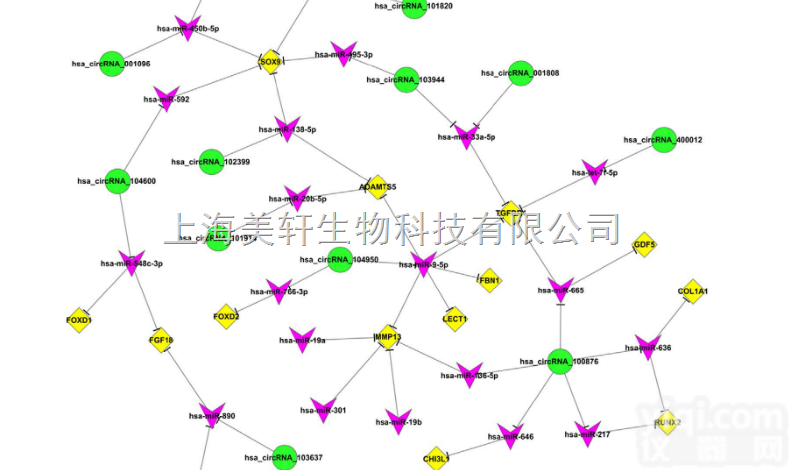
6 miRNA 结合位点分析
circRNA 可以通过与 miRNA 结合的方式YZ miRNA 的功能(Thomas B. Hansen et al., 2013)。因此,对鉴定的 circRNA 进行 miRNA 结合位点分析,有助于进一步研究 circRNA 的功能。对于动物和植物,分别使用 miRanda 和 psRobot 软件预测剪切后的 circRNA 的 miRNA 的结合位点。部分结果
见下表:
hsa-let-7a-2-3p hsa_circ_0000020
hsa-let-7a-2-3p hsa_circ_0000021
hsa-let-7a-2-3p hsa_circ_0000022
hsa-let-7a-2-3p hsa_circ_0000023
hsa-let-7a-2-3p hsa_circ_0000024
hsa-let-7a-2-3p hsa_circ_0000027
hsa-let-7a-2-3p hsa_circ_0000028
hsa-let-7a-2-3p hsa_circ_0000029
hsa-let-7a-2-3p hsa_circ_0000041
hsa-let-7a-2-3p hsa_circ_0000050
 环状RNA测序
环状RNA测序
 circRNA/环状RNA测序
circRNA/环状RNA测序
 环状RNA测序
环状RNA测序
 circRNA 环状RNA测序
circRNA 环状RNA测序
 环状RNA测序
环状RNA测序
 circRNA(环状RNA)测序分析服务
circRNA(环状RNA)测序分析服务
 CleanNA CleanNGS 高通量测序文库DNA/RNA纯化及片段筛选磁珠
CleanNA CleanNGS 高通量测序文库DNA/RNA纯化及片段筛选磁珠
 环状RNA(circRNA)测序技术服务
环状RNA(circRNA)测序
环状RNA(circRNA)测序技术服务
环状RNA(circRNA)测序
 Small RNA测序/小RNA测序/microRNA测序/miRNA测序/lncRNA测序
Small RNA测序/小RNA测序/microRNA测序/miRNA测序/lncRNA测序
 Small RNA测序/小RNA测序/microRNA测序/miRNA...
Small RNA测序/小RNA测序/microRNA测序/miRNA...
 转录组测序//RNA seq//16s 扩增子测序//全基因组测序//全外显子组测序//单细胞测序//高通量测序//NGS
转录组测序//RNA seq//16s 扩增子测序//全基因组测序//全外显子组测序//单细胞测序//高通量测序//NGS
本产品信息由(北京中康博生物科技有限公司)为您提供,内容包括(环状RNA测序 )的品牌、型号、技术参数、详细介绍等;如果您想了解更多关于(环状RNA测序 )的信息,请直接联系供应商,给供应商留言。若当前页面内容侵犯到您的权益,请及时告知我们,我们将马上修改或删除。








